El experimento para fármaco cuya venta no está autorizada se realiza en siete hospitales
En Hospital Barros Luco intentan experimentar fármaco de Laboratorio Merck con paciente analfabeta
21.08.2013







Hoy nuestra principal fuente de financiamiento son nuestros socios. ¡ÚNETE a la Comunidad +CIPER!
El experimento para fármaco cuya venta no está autorizada se realiza en siete hospitales
21.08.2013




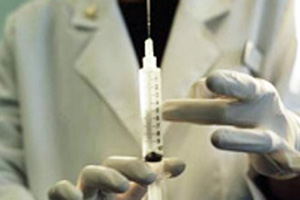
Cora Gajardo ingresó a la sala J de la unidad de Medicina Interna del Hospital Barros Luco y se sentó en una silla junto a la cama de Rosa Lizana, su consuegra. Era el mediodía del martes 9 de julio, la hora de almuerzo, y como Rosa a duras penas podía sentarse en su camilla, Cora le ayudaría a comer y le haría compañía. Para entonces, Rosa llevaba poco más de un mes internada en ese hospital. No por la insuficiencia renal que desde hace siete años la obliga a someterse a diálisis tres veces por semana, sino por una infección vesicular. Tenía dolores estomacales, indigestión, vómitos. Apenas la internaron, los médicos le practicaron una intervención quirúrgica menor para evacuar la bilis y estabilizarla. Todo salió bien, pero pronto le diagnosticaron una infección respiratoria, la que el equipo médico no demoró en controlar. Pero luego se le obstruyó la fístula con la que se dializa. Fue entonces que todo se complicó.
Porque Rosa Lizana, de 74 años, contrajo una infección intrahospitalaria causada por la bacteria clostridium difficile, un microorganismo que puede producir infecciones intestinales provocando diarrea severa, colitis seudo membranosa, megacolon tóxico, perforación del colon y, en los casos más severos, la muerte.
 Su perfil clínico la ubicaba entre la población de más alto riesgo: mayor de 65 años y con una patología renal. Pero también la convertía en candidata perfecta para participar como sujeto de estudio en un ensayo clínico que el laboratorio estadounidense Merck & Co. Inc. realiza en Chile desde comienzos de 2012 para poner a prueba tres drogas experimentales que atacan las toxinas del clostridium difficile: los anticuerpos MK-3415, MK-6072 y MK-3415A. El estudio, que según los registros de la Food and Drugs Administration (FDA) de Estados Unidos se lleva a cabo simultáneamente en otros 18 países, pretende determinar la eficacia, seguridad y tolerabilidad de los fármacos en pacientes mayores de 18 años, cuyo diagnóstico haya sido detectado durante la última semana y que se encuentren bajo tratamiento estándar con antibióticos.
Su perfil clínico la ubicaba entre la población de más alto riesgo: mayor de 65 años y con una patología renal. Pero también la convertía en candidata perfecta para participar como sujeto de estudio en un ensayo clínico que el laboratorio estadounidense Merck & Co. Inc. realiza en Chile desde comienzos de 2012 para poner a prueba tres drogas experimentales que atacan las toxinas del clostridium difficile: los anticuerpos MK-3415, MK-6072 y MK-3415A. El estudio, que según los registros de la Food and Drugs Administration (FDA) de Estados Unidos se lleva a cabo simultáneamente en otros 18 países, pretende determinar la eficacia, seguridad y tolerabilidad de los fármacos en pacientes mayores de 18 años, cuyo diagnóstico haya sido detectado durante la última semana y que se encuentren bajo tratamiento estándar con antibióticos.
Desde su silla, Cora Gajardo vio que su consuegra tenía una vía intravenosa puesta en el brazo, algo que no había visto en ninguna de sus visitas previas. “Me van a dar un nuevo remedio”, le dijo Rosa. Cora le preguntó de qué se trataba, pero Rosa le respondió que no sabía muy bien, que durante esa misma mañana había llegado una doctora y le había hecho firmar un papel, y que sólo le dijo que le cambiarían el tratamiento. El documento con su firma estaba a los pies de Rosa, sobre su cama. Llevaba por título “Forma de Consentimiento”. Cora lo tomó y comenzó a leerlo en voz alta:
“Se le invita a participar en un estudio de investigación. Este formato de consentimiento contiene información que le ayudará a decidir si desea participar. Tome su tiempo, lea este formato atentamente y formule a su médico o personal del estudio cualquier pregunta que pueda tener. Usted no debe firmar este formato hasta que haya entendido toda la información contenida en las siguientes páginas y hasta que todas sus preguntas acerca de la investigación hayan sido respondidas satisfactoriamente. El médico del estudio será pagado por el Patrocinador, Merck Sharp & Dohme Chile por la conducción de este estudio”.
A medida que Cora leía, el rostro de Rosa iba cambiando. Estaba aterrada. Entonces Cora se detuvo, pero siguió hojeando en silencio las 14 páginas del documento. Vio que se trataba de fármacos que aún no han sido aprobados para su venta. También supo que, en todo el mundo, serían unas 1.600 personas las que participarían en el estudio y que, si Rosa aceptaba, se arriesgaba a sufrir una serie de posibles efectos secundarios producto de las drogas experimentales, sin siquiera tener garantías de que curaría su enfermedad. Algunos estaban enunciados: malestar estomacal, nauseas, diarrea, vértigo, baja en la presión arterial, cansancio, dolor de cabeza, pérdida de apetito, erupción cutánea, inflamación del colon, disminución de los niveles de sodio en la sangre, infección de las vías urinarias, vómitos y heces con sangre, entre otros tantos.
Pero al final de todo, en la última hoja, Cora vio lo que le pareció más grave: la firma de Rosa Lizana. ¿Por qué lo más grave? Porque Rosa no sabe leer ni escribir, salvo su nombre.
A comienzos de 2012, el laboratorio británico GlaxoSmithKline y dos de sus investigadores fueron sentenciados por un tribunal argentino a pagar una multa de US$ 230.000. Aunque el monto parece irrisorio para una compañía que ocupa el segundo lugar en el ranking de las mayores empresas farmacéuticas a nivel mundial –superado sólo por la estadounidense Pfizer–, el fallo generó alarma porque abrió una ventana al sistema utilizado por la industria para reclutar sus sujetos de estudio en distintas partes del planeta, enfocándose principalmente en pacientes de los estratos más vulnerables.
La multa se originó luego de que la Administración Nacional de Medicamentos de Argentina (ANMAT) detectara irregularidades en un estudio que GlaxoSmithKline realizó en ese país entre 2007 y 2008. El experimento que llevaba por nombre COMPAS (ver información del estudio), consistió en administrar una vacuna experimental –el fármaco GSK1024850A– para prevenir la neumonía a cerca de 14.000 niños menores de 3 años en tres provincias de ese país. Aunque los resultados del ensayo clínico no fueron cuestionados, sí lo fue la forma en que el gigante farmacéutico reclutó a los pacientes: los padres de varios de esos niños eran analfabetos y no comprendían el texto del denominado «consentimiento informado«, el formulario que se firma aceptando las condiciones de las pruebas médicas. Por lo tanto, nunca supieron el potencial riesgo que corrían sus hijos al participar en el estudio. 14 niños murieron tras la inoculación de esa vacuna. Nunca quedó clara la causa de muerte.
 La misma falta de información al paciente ocurrió a comienzos de julio pasado en el Hospital Barros Luco. Sin saberlo, con su firma, Rosa Lizana había accedido “voluntariamente” a participar en uno de los 142 ensayos clínicos abiertos que están reclutando pacientes en Chile (ver estudios). Todos esos estudios aparecen registrados en el sitio Clinicaltrials.gov, el portal de acceso público donde la FDA, la autoridad sanitaria de Estados Unidos, registra los proyectos que los laboratorios farmacéuticos realizan en cualquier país del mundo para poner a prueba fármacos experimentales.
La misma falta de información al paciente ocurrió a comienzos de julio pasado en el Hospital Barros Luco. Sin saberlo, con su firma, Rosa Lizana había accedido “voluntariamente” a participar en uno de los 142 ensayos clínicos abiertos que están reclutando pacientes en Chile (ver estudios). Todos esos estudios aparecen registrados en el sitio Clinicaltrials.gov, el portal de acceso público donde la FDA, la autoridad sanitaria de Estados Unidos, registra los proyectos que los laboratorios farmacéuticos realizan en cualquier país del mundo para poner a prueba fármacos experimentales.
El Laboratorio Merck, patrocinador del estudio de los anticuerpos para atacar al clostridium difficile, es responsable de 14 de los ensayos clínicos que están actualmente vigentes en Chile a través de su subsidiaria: Merck Sharp & Dohme I.A. Corp. Así, se ha convertido en la segunda empresa farmacéutica con mayor actividad de investigación en el país, antecedida sólo por la suiza Hoffmann-La Roche (16 ensayos) y seguida por GlaxoSmithKline (12 ensayos).
Su primer acercamiento para realizar ese estudio en el Hospital Barros Luco fue con el doctor Carlos Beltrán, jefe de la Unidad de Infectología, quien ya había participado como investigador en varios ensayos clínicos con distintos laboratorios, principalmente en pacientes con VIH. Pero esta vez no quiso hacerlo. Recomendó a su equipo para que lo llevara adelante bajo la conducción de la doctora Pilar Gambra. Ella fue la primera profesional que se hizo cargo del estudio en el hospital cuando comenzó a aplicarse en el país a comienzos de 2012. Pero un año y medio después, cuando Rosa Lizana firmó el documento, Gambra ya había dejado de trabajar allí. Para entonces, el ensayo clínico estaba en manos de la doctora Sofía Palma, la infectóloga que hasta hoy es la investigadora responsable del proyecto en ese centro asistencial.
Habían pasado sólo unos minutos desde que Cora Gajardo leyera el documento que su consuegra Rosa Lizana firmó esa misma mañana cuando una enfermera entró a la sala J del Hospital Barros Luco. Traía en sus manos un preparado que se dispuso a conectar a la vía intravenosa que Rosa tenía puesta en su brazo. Cora ya había llamado al hijo de Rosa, Mauricio Vargas, informándole del formulario que su madre había suscrito sin saber lo que firmaba. En ese minuto, él ya iba camino al hospital. Cora relató a CIPER que en ese momento le preguntó a la enfermera qué era lo que le administrarían a Rosa y que la mujer le respondió que era la droga que aparecía en la “forma de consentimiento”. Cora le impidió que lo hiciera sin que estuviera su yerno presente y sin que antes le explicaran de qué trataba todo eso.
Lo extraño es que, formalmente, la enfermera no debía inyectarle nada a Rosa. Porque, según explicó a CIPER la doctora Sofía Palma, el proceso de “consentimiento informado” todavía no había concluido y, por lo tanto, Rosa aún no era oficialmente parte del estudio.
El estudio que dirige Palma y en el que Rosa estaba siendo enrolada, es de fase III, lo que significa que está en su última etapa de investigación antes de poder salir al mercado. Además, es “doble ciego”, aleatorio y controlado por placebo. En otras palabras, ni el médico a cargo ni el paciente saben cuál de los fármacos del estudio se está aplicando, ya que la mezcla que se le administra al paciente es determinada al azar en Estados Unidos. La información llega a Chile a través de un sistema computacional para que una enfermera “ciega” –que no tiene contacto directo con el paciente– prepare la droga. Por lo tanto, las personas que participan del estudio pueden ser inoculadas por vía intravenosa con el anticuerpo MK-3415 (que ataca la toxina A del clostridium difficile), el MK-6072 (que ataca la toxina B), ambos a la vez (MK-3415A, contra las dos toxinas) o un placebo compuesto por una solución salina sin ingredientes activos. El procedimiento, que está siendo aplicado en siete hospitales públicos del país, pretende demostrar la eficacia de los nuevos fármacos usados en forma paralela a la terapia estándar con antibióticos (vancomicina y metronidazole). El objetivo es disminuir la recurrencia del clostridium difficile, ya que en el 20% de los casos la infección vuelve una vez que los pacientes han sido dados de alta.
Los siete hospitales chilenos con los cuales el Laboratorio Merck se asoció para realizar el experimento de los anticuerpos contra la infección intrahospitalaria provocada por la bacetria clostridium difficile, son: la Posta Central y los hospitales Barros Luco, San Borja Arriarán, Sótero del Río, Padre Hurtado, en Santiago; el Hospital Gustavo Fricke, en Viña del Mar y el Hospital de Valdivia, en la Región de Los Ríos.
Chile se ha convertido en los últimos años en un destino de alta demanda para la experimentación científica en seres humanos. De acuerdo a los registros del Instituto de Salud Pública (ISP), el estudio del laboratorio Merck es sólo uno de los 858 ensayos clínicos que la autoridad sanitaria ha permitido que se realicen en el país desde 2006 (ver base de datos). La ley que regula la investigación médica, a siete años de su promulgación, aún tiene tan sólo tres páginas y presenta varios vacíos (ver recuadro). Aún así, para proteger a los pacientes de posibles abusos, es clara al establecer como regla básica que “toda investigación científica en un ser humano deberá contar con su consentimiento previo, expreso, libre e informado, o, en su defecto, el de aquel que deba suplir su voluntad en conformidad con la ley”.
 El mismo principio está incluido en la Guía de Buenas Prácticas Clínicas, el conjunto de normas internacionales que rigen la investigación científica en personas a nivel mundial. Por eso, cuando el ingeniero Mauricio Vargas –hijo de Rosa Lizana- llegó al hospital y leyó el documento que su madre había firmado, no sólo se asustó sino que también se enfureció.
El mismo principio está incluido en la Guía de Buenas Prácticas Clínicas, el conjunto de normas internacionales que rigen la investigación científica en personas a nivel mundial. Por eso, cuando el ingeniero Mauricio Vargas –hijo de Rosa Lizana- llegó al hospital y leyó el documento que su madre había firmado, no sólo se asustó sino que también se enfureció.
-Mi mamá es analfabeta y no asocia los números con los meses, así que si escribió junto a su firma la fecha “090713”, fue porque se lo dictaron. ¡Si hasta los 40 años nunca ingresó a un colegio! No sabe sumar, restar, multiplicar, dividir, leer ni escribir. Si le pasas cualquier libro en español, alemán, inglés, rumano o francés, para ella es lo mismo: no entiende nada. No está en condiciones de leer ni menos comprender el documento que le hicieron firmar –afirmó a CIPER.
Mauricio Vargas pidió hablar de inmediato con los médicos. Primero lo hizo con el jefe de la sala del Hospital Barros Luco donde Rosa estaba internada. Luego con Loreto Rojas, una de las tres doctoras que forman parte del equipo a cargo del estudio. Fue ella quien se acercó a Rosa la mañana del 9 de julio con el formulario de consentimiento. De hecho, su firma también está en la última página del documento de 14 páginas. La doctora Rojas aseguró a CIPER que sí le preguntó a Rosa si sabía leer y escribir y que ella le respondió que «sólo escribía con letra imprenta». También señaló que le explicó «detalladamente de qué trataba el estudio, ella aceptó y luego firmó el consentimiento de forma totalmente voluntaria». Y que además, «se le contestaron todas sus dudas». Eso fue lo que trató de explicarle a Mauricio, pero él estaba demasiado alterado para escuchar. Según la doctora Rojas, «se comportó de una manera muy descortés, con un descontrol absoluto».
En todo caso, Mauricio tenía motivos para alterarse. Apenas leyó el documento, le preguntó a su madre y confirmó que no tenía idea de qué había firmado. Pero además, todo había ocurrido en pocos minutos en la sala donde Rosa estaba internada, por lo que era imposible que la doctora Rojas le hubiera explicado el contenido de las 14 páginas del documento donde aparece su firma al final. Como Mauricio no estaba conforme con las explicaciones de la doctora Loreto Rojas, exigió hablar con la conductora del ensayo, la doctora Sofía Palma.
Palma relató a CIPER que intentó explicarle a Mauricio que, en el caso de Rosa Lizana, el proceso de consentimiento informado no se completó: “Nosotros hablamos con él. Pero pensó que su mamá ya estaba incluida porque había puesto su firma en el consentimiento. Sin embargo, ella tenía un tutor legal y en definitiva, el proceso de consentimiento informado no se concluyó, así que la paciente no entró al estudio”.
-¿Qué era lo que faltaba para ser incluida? –le preguntamos.
-Faltó la firma del ministro de fe y además, en este caso, ella tenía un tutor legal que también debía firmar.
El reglamento de la Ley 20.120 establece que en cualquier ensayo clínico, el consentimiento informado “deberá constar en un acta firmada por la persona que ha de consentir en la investigación, por el investigador responsable o principal, en su caso, y por el director del centro o establecimiento donde ella se llevará a cabo, quien, además, actuará como ministro de fe”. Por lo tanto, en estricto rigor, para que Rosa fuera enrolada en el estudio, faltaba la firma del director del Hospital Barros Luco, el doctor Luis Leiva, quien tiene el mandato legal de actuar como garante de que la persona que firma el consentimiento lo haga de forma voluntaria e informada. Y como Rosa tenía un tutor legal (su hija Marcela Vargas), también faltaba su firma.
Según la doctora Palma, fue la misma paciente quien les dijo que sus familiares llegarían esa tarde, y fue por eso que dejaron junto a ella una copia del documento: para que lo revisaran juntos durante el tiempo que sea necesario. Que por eso, el proceso de consentimiento informado a veces puede tardar días en concretarse. Además, aseguró que para este estudio, el apoyo de la familia es fundamental, ya que durante los tres meses que dura la investigación, el paciente debe escribir a diario un registro detallado con el estado de sus deposiciones y su temperatura. Algo que Rosa no habría podido hacer por sí sola.
A pesar de las explicaciones de la encargada del estudio, hay varias cosas que no cuadran. Para que el director del hospital –o su representante– pudiera garantizar que Rosa Lizana accedía de forma “voluntaria” e “informada” a participar en el estudio, debió haber estado allí, pero eso no ocurrió. Tampoco estaba su tutora legal. Palma reconoce que no haberla esperado ni haberla llamado fue un error. Pero tampoco explica por qué se permitió que una mujer analfabeta firmara el documento. Y menos, por qué la enfermera llegó ese mismo día con los fármacos experimentales para administrárselos a Rosa. De ahí se puede deducir que, finalmente, las firmas que faltan en el papel no son más que un tecnicismo, una formalidad, porque la más importante, la de la paciente que accede a someterse al estudio –como sucedió con Rosa Lizana-, ya estaba puesta en la última página del documento.
En noviembre de 2012, la ONG Políticas Farmacéuticas publicó un breve pero completo informe sobre la industria de los ensayos clínicos en el país. Allí aparecen datos interesantes: como que Chile es la nación que concentra la mayor cantidad de pruebas clínicas per cápita en la región, que el 70% de los estudios que se realizan en el país está concentrado en sólo 10 laboratorios multinacionales, que apenas un 4,4% son patrocinados por entidades chilenas y que ese porcentaje se compone principalmente de universidades, ya que los laboratorios nacionales no invierten en la investigación de nuevos fármacos. En ese sentido, el informe concluye: “La información que se genera en los pacientes chilenos termina principalmente siendo ocupada para la explotación de una patente de un laboratorio extranjero”.

Fuente: Clinicaltrials.gov
El doctor en Farmacología y director de contenidos de la ONG Políticas Farmacéuticas, Rodrigo López, asegura que son dos los principales factores que han convertido a Chile en un atractivo lugar para realizar ensayos clínicos: la calidad técnica de los profesionales chilenos y los pacientes que se encuentran principalmente en los hospitales del país.
-Los laboratorios buscan países donde los costos sean relativamente bajos, donde haya pacientes para las patologías y donde el dato que se extraiga de la investigación sea confiable. Esa combinación de factores ha hecho que Chile sea un buen lugar para hacer estudios clínicos. En ese sentido, también es importante la cantidad de comorbilidad (otras patologías) que pueden tener los sujetos de estudio: en Chile, de cierta forma, el paciente está un poquito más limpio. En África u otros países de Latinoamérica puede ser más barato, pero la posibilidad de encontrarte con infecciones parasitarias y otras enfermedades es mayor, por lo que produce mucha variabilidad de los datos. El estado de salud del chileno promedio también influye en que el país sea un buen lugar para realizar los estudios –dice López.
Si se considera que un estudio del Manhattan Institute for Policy Research indicó que los laboratorios internacionales invierten en promedio más de US$ 1.300 millones por cada investigación de un nuevo fármaco, y que el 90% de ese presupuesto se destina a los estudios de fase III, que son la mayoría de los que se realizan en Chile, ese mercado ha generado un nuevo y lucrativo nicho de negocio para los profesionales chilenos que participan en este tipo de proyectos.
El doctor Harold Mix, director del Diplomado en Investigación Clínica y Monitoreo de Ensayos de la Universidad de Chile, explica que el pago a los investigadores y sus equipos varía dependiendo del estudio, pero que es directamente proporcional al tiempo que dedican a la investigación y que la tarifa es muy similar a lo que se cobra en las clínicas caras de Santiago.
-Si un médico de estas clínicas ve a tres o cuatro pacientes en una hora, y a cada uno le cobra más de $50.000 por la consulta, saca el cálculo de cuánto gana ese médico al dedicar esa hora a la investigación. Lo que se paga es el tiempo, y no sólo al investigador, sino que a todas las personas que participan. Si hay una enfermera que tomó los exámenes, se le paga en proporción. Y todos los exámenes también están contemplados en el presupuesto –dice Mix.
Tomando en cuenta que en esas clínicas el sueldo puede llegar a triplicar lo que gana un médico en el sector público, y que la mayor parte de los estudios se llevan a cabo en hospitales, el negocio para los investigadores resulta redondo.
Según la información disponible en su sitio web, en el Hospital Clínico de la Universidad de Chile hay en curso 20 estudios clínicos patrocinados por laboratorios internacionales. La coordinadora de todos esos ensayos es una química farmacéutica. Un profesional que ha participado en varios de esos estudios cuenta que ha visto proyectos cuyo presupuesto varía entre US$ 1.000 y US$ 18.000 por cada paciente enrolado, lo que no significa que ese dinero vaya a parar a los bolsillos del equipo clínico a cargo del estudio. De ese fondo se descuentan los gastos en exámenes y el overhead que algunos establecimientos –no todos- cobran por el uso de sus instalaciones y que por lo general consiste entre el 10% y el 20% de lo que recibe el equipo investigador. En todo caso, si se considera que hay estudios que contemplan el enrolamiento de casi un centenar de pacientes, la cifra no es nada despreciable.
Mientras eso ocurre, los pacientes no resultan necesariamente beneficiados. En el caso del ensayo del laboratorio Merck donde Rosa Lizana estuvo a punto de ser incluida, se espera que por cada uno de los siete hospitales donde se está llevando a cabo se recluten unos 10 sujetos de estudio. Salvo los gastos de movilización y la gratuidad de los controles y exámenes necesarios para que los investigadores puedan recopilar la información, ninguno recibirá remuneración alguna por participar. Sólo obtendrán un beneficio si las drogas que probaron en ellos resultan ser efectivas. Pero incluso ese beneficio se acabará cuando el estudio termine: no hay ninguna norma que les garantice el derecho a continuar con el tratamiento una vez que salga al mercado.
-La mayoría de los estudios clínicos se hacen en los hospitales con pacientes con bajo nivel de acceso a salud de alto costo. Pero el medicamento que resulta de ese estudio sale al mercado con un precio altísimo, permitiéndole el acceso a otro grupo de la población muy distinto. La población que se somete a los estudios está sujeta a un riesgo, alto o bajo, da lo mismo, pero existe. Y es muy poco probable que reciba parte del beneficio. Esa es la lógica que hay que equilibrar –dice Rodrigo López.
De ese “equilibrio” la familia de Rosa Lizana hoy sabe bastante. Todos los abogados que han consultado por lo ocurrido con la mujer de 74 años, que estuvo a punto de ser inoculada con un medicamento en fase experimental, han recomendado desistir de toda acción judicial. “Si no hay daños físicos, no hay juicio”.
La experimentación de fármacos en seres humanos es una práctica que se ha desarrollado en Chile, al menos, desde la década de los ’80. Por lo tanto, la presencia de grandes laboratorios internacionales en el país no es reciente. Lo que sí es nuevo son las normas chilenas que regulan la actividad.
Recién en 2001 el Minsal publicó la Norma Técnica Nº 57, el primer documento que regula la ejecución de ensayos clínicos en el país. Allí se establecen los requisitos que deben cumplir investigadores, laboratorios y las instituciones donde se llevan a cabo las pruebas. También el rol del ISP como entidad reguladora de los fármacos sin registro en el país que se someten a estudio. Aunque es también el primer documento que se refiere a la protección de los pacientes que participan en ensayos clínicos, como es sólo una norma técnica, ese resguardo no estaba garantizado, ya que no era materia de ley. Tuvieron que pasar otros cinco años para que la legislación tomara en cuenta a los cientos de pacientes que cada año se someten a tratamientos con drogas experimentales.
La misma ley que prohíbe la clonación y las prácticas eugenésicas en Chile es la que regula la investigación bioética en el país (Ley Nº 20.120). En sólo tres páginas establece, entre otras cosas, los requerimientos básicos para que la industria farmacéutica ponga a prueba nuevos medicamentos en pacientes chilenos. Algunos de ellos son: que todos los estudios deberán ser aprobados por el director de cada establecimiento, previo informe favorable de los comités de ética que corresponda; que no se aprobarán tratamientos experimentales “si hay antecedentes que permitan suponer que existe un riesgo de destrucción, muerte o lesión corporal grave y duradera para un ser humano”; y que toda persona que participe como sujeto de estudio debe hacerlo voluntariamente mediante un formulario de consentimiento informado.
Para su cumplimiento, el mismo cuerpo legal establece que se regirá por un reglamento. El problema es que ese reglamento no apareció sino hasta 2011 (ver reglamento), por lo que durante los primeros cinco años que la ley estuvo vigente, la mayoría de los aspectos vinculados a los ensayos clínicos y su práctica en Chile estuvo desregulada.
La ley de 2006 también estableció la creación de una Comisión Nacional de Bioética que asesoraría “a los distintos Poderes del Estado en los asuntos éticos que se presenten como producto de los avances científicos y tecnológicos en biomedicina, así como en las materias relacionadas con la investigación científica biomédica en seres humanos, recomendando la dictación, modificación y supresión de las normas que la regulen”. Esa comisión nunca se constituyó.
Recién en octubre de 2012, el ministro de Salud, Jaime Mañalich, anunció la creación de CEMEIS, la nueva Comisión Ministerial de Ética de la Investigación en Salud, cuyos objetivos principales serán establecer parámetros homogéneos y acreditar la idoneidad de las comisiones éticas regionales en la investigación clínica, para así generar una política universal de Consentimiento Informado para los Pacientes. Está integrada por Gladys Borquez, magister en Bioética y académica de la Facultad de Medicina de la Universidad de Chile; María Inés Gómez, profesora del Centro de Bioética de la Facultad de Medicina de la Universidad del Desarrollo; Juan Lecaros, abogado especialista en bioética; Helen Rosenbluth, subjefa de ANAMED del Instituto de Salud Pública y Francisco León, presidente de la Sociedad Chilena de Bioética.








